
・Letter to the Editor・
Dominant
cystoid macular dystrophy associated with mutations in the RP
Yan Fu1, Tian-Hao Xie2, Yue-Ling
Zhang1, Na Yang1, Xiao-Nan Shi1, Zhao-Hui Gu1
1Department of Ophthalmology, Baoding
First Central Hospital, Baoding 071000, Hebei Province, China
2Affiliated Hospital of Hebei
University, Baoding 071002, Hebei Province, China
Correspondence to: Zhao-Hui Gu. Department of Ophthalmology,
Baoding First Central Hospital, Baoding 071000, Hebei Province, China.
zhaohui-gu@sohu.com
Received:
DOI:10.18240/ijo.2019.12.23
Citation:
Fu Y, Xie TH, Zhang YL, Yang N, Shi XN, Gu ZH. Dominant cystoid macular
dystrophy associated with mutations in the RP
Dear Editor,
We describe in detail a case of
dominant cystoid macular dystrophy (DCMD) patient carrying a novel heterozygous
RP
Our study included four family
members from three generations. The research protocol was approved by the
Ethical Committee of Baoding First Central Hospital (Hebei Province, China).
Informed written consent was provided by all participants following a detailed
explanation of the procedures. Blood samples were collected from the patient
and her brother and genomic DNA was extracted from peripheral blood lymphocytes
for genetic testing and subsequent analysis.
A 37-year-old female patient
(proband, P3) presented with a 5-month history of visual dimness in both eyes
without other discomfort (e.g., photopsia, diplopia, night blindness,
eye pain) or systemic symptoms (e.g., headache). She had
non-contributory family history. Her medical and social histories were
unremarkable. Although subjectively declined, the best corrected visual acuity
(BCVA) remained 1.0 and
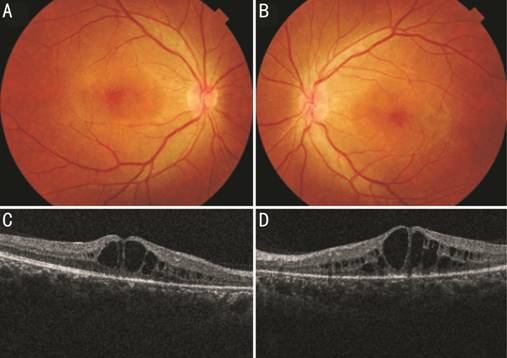
Figure 1 Color fundus photograph of
right (A) and left (B) eye showing cystoid macular edema at posterior pole.
Spectral domain optical coherence tomography line scan of right (C) and left
(D) eye shows schisis at the inner nuclear layer and outer nuclear layer/outer
plexiform layer involves the entire macular area.
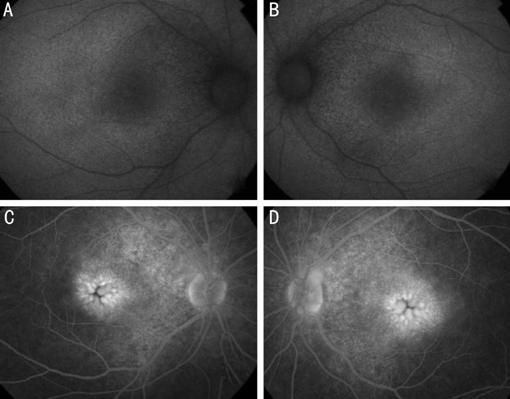
Figure 2 Autofluroscence of right
(A) and left (B) eye showing very faint hyperautofluorescence surrounding the
fovea involving the posterior pole extending within the arcades. Fundus
fluoresce in angiography latephase of right (C) and left (D) eye showing very
petaloid hyperfluorescence at the fovea, note the hyperfluorescence of the area
around the disks. The retinal pigment epithelial has an indistinct granular
hypopigmented changes.
The family history of this patient
was remarkable. She was the second child born to non-consanguineous Chinese
parents. Her mother (P1), committed suicide in her thirties. The patient stated
that her father (P2) and brother (P4) had a similar history of vision loss.
Four family members, including the proband, her father (57 years old), older
brother (39 years old), and nephew (9 years old) participated in the study. The
clinical characteristics of the four individuals are shown in Table 1. Her
brother first noted poor central vision at the age of approximately 30y. He was
diagnosed with bilateral CME at the age of 34y. His BCVA was 0.2 and
Table 1 Clinical characteristics of
the proband and family members
|
Patients |
P3 |
P4 |
P2 |
P6 |
|
Age (y) |
37 |
39 |
57 |
9 |
|
Onset (y) |
37 |
34 |
Middle age |
- |
|
BCVA (OD/OS) |
1.0/0.4 |
0.2/0.6 |
0.2/0.2 |
1.0/1.0 |
|
Fundus |
Macular edema |
Macular edema |
Macular atrophy |
Normal |
|
OCT |
CME in both eyes |
CME in both eyes |
Disruption and loss of the ellipsoid zone |
Normal |
|
FFA |
Petaloid leakage at the fovea in both eyes |
Petaloid leakage at the fovea in both eyes |
- |
- |
|
FAF |
Normal |
Normal |
- |
- |
|
Color vision test |
Normal |
Normal |
Normal |
Normal |
BCVA: Best corrected visual acuity;
OD: Right eye; OS: Left eye; OCT: Optical coherence tomography; FFA: Fundus fluorescein
angiography; FAF: Fundus autofluroscence; CME: Cystoid macular edema.
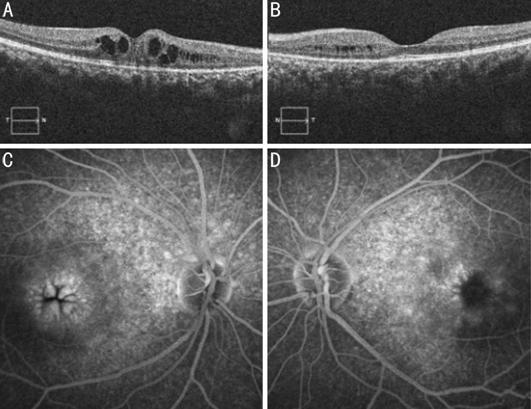
Figure 3 Spectral domain optical coherence
tomography showed cystoid spaces in both eyes (A: right eye; B: left eye).
Fluorescein angiography study demonstrating the typical petalloid pattern
observed in cystoid macular edema (C: right eye; D: left eye).
A diagnosis of DCMD was reached based
on the history, clinical examination, and imaging findings. The proband’s father had a visual acuity of
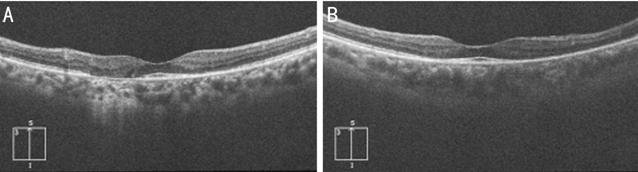
Figure 4 Optical coherence
tomography showed disruption and loss of the ellipsoid zone, nodular elevations
on retinal pigment epithelium. The photoreceptor inner segment/outer segment
junction line was intact at the macula in right (A) and left (B) eye.
Informed consent for genetic
analysis was provided by the proband and her brother. In our study, we
comprehensively screened all 381 genes associated with common eye diseases,
identifying three mutations in the RP
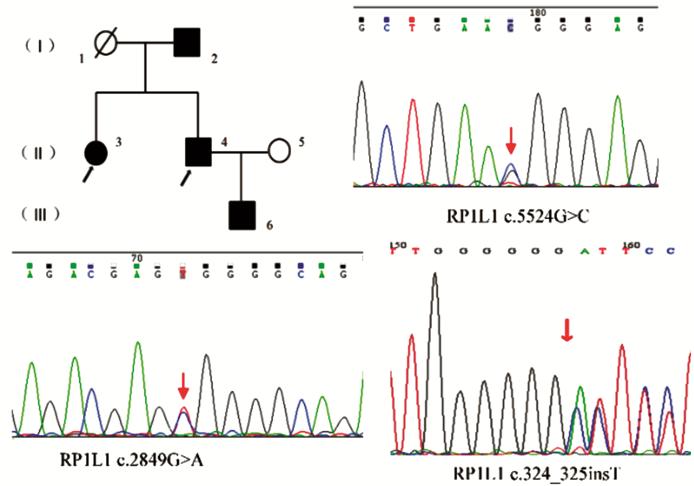
Figure 5 Pedigree and identified mutations
of a family with DCMD Affected patient is shown with a
solid symbol and unaffected with open symbols. Black arrows: Genotype analysis
performed; Squares: Male; Circles: Female; Slashed symbols: Deceased; Red
arrows: The position of the mutated nucleotide.
DISCUSSION
DCMD is an inherited autosomal
dominant disease causing an early onset of cystic edema in the posterior pole
of the eye. Pinckers et al[1] first
described the retinal functinon in DCMD, while Saksens et al[2] reported the clinical features and long-term follow-up
outcome of the disease. In many different hereditary macular dystrophies, CME
exhibits a specific pattern of presentation. Roy et al[3]
described the multimodal imaging features in a case with DCMD. In the present
article, we report a family affected by this dystrophy and note its clinical
characteristics. We performed genetic testing and comprehensive mutation
analysis, and demonstrated that the RPIL1 gene may be responsible for
the retinal phenotype.
This is a case of a female patient
who had bilateral visual loss and remarkable early onset of CME in her 30
years. Her father and brother had a history of bilateral visual loss. Her
brother having similar fundus findings. The brother and sister had CME at the
posterior pole. Otherwise, there were no evident abnormalities in the fundus.
Spectral domain optical coherence tomography revealed cystoid spaces. FA showed
the typical characteristics of petaloid hyperfluorescence leakage. The ERG was
normal.
Several other diagnoses with
early-onset CME should be considered in this clinical setting, such as
anterior, intermediate and posterior uveitis[4], retinal
vascular disease, intra-ocular surgery history, vitreo-macular traction
syndrome, age-related macular degeneration, following cataract extraction,
choroidal tumors, toxic retinopathy, X-linked juvenile retinoschisis (XLRS),
and retinitis pigmentosa.
The patient had no history of
uveitis. Peripheral fundus examination was performed, and biomicroscopy showed
that the anterior vitreous was clear. There was no evidence of pars planitis
and posterior choroiditis. Several vascular retinal abnormalities have been
related to the development of CME (e.g., Coat’s disease, central or
branch retinal vein occlusion[5], diabetic
retinopathy, and perifoveal retinal telangiectasis). The patient did not have
an underlying systemic disorder that could predispose her to ocular retinal
vascular occlusion or inflammation. Specifically, there were no clinical signs
of retinal ischemia or neovascularization. Choroidal tumors, such as choroidal
hemangioma and choroidal melanoma, are occasionally associated with CME. There
was no evidence of choroidal tumors. Various retinal toxicities associated with
CME have been described for agents such as tamoxifen, nicotinic acid, and
epinepherine. This condition is reversible and the macular cyst will resolve following
the cessation of drug intake[6]. The patient
denied using any medication, and did not mention vitreo-retinal traction or
prior intra-ocular surgery.
XLRS, which is inherited in an
X-linked pattern compared with DCMD, is frequently diagnosed prior to school
age, indicating a juvenile onset. The XLRS is marked by the typical presence of
a spoke-wheel pattern in the macula of patients aged <30y. The cystoid
spaces are mainly situated in the inner and outer nuclear layers, without
characteristic fluorescein leakage into the cystoid schisis cavities, as shown
on FA[7]. The distinctive ERG symbol is a
‘negative’ ERG caused by a bright flash of light in the dark-adapted retina, in
which the a-wave is larger than the b-wave as opposed to DCMD. Retinitis
pigmentosa should also be considered[8]. Patients
with retinitis pigmentosa exhibit bone-spicule pigmentary changes, arteriolar
attenuation in the early stages of the disease and early experience of night
blindness in contrast to those with DCMD.
The hallmark of family history is
that both the older brother and father of the proband had poor vision. The
presence of typical clinical features led to the diagnosis of DCMD. Thus far,
there are no effective treatments for DCMD. Intramuscular long-acting
octreotide acetate appeared effective in the stabilization of visual acuity[9]. The later stages of DCMD manifest as macular atrophy.
The progression of the disease is accompanied by reduction in the cystic
spaces, giving rise to chorioretinal atrophy with subsequent loss of vision.
The patient’s father may be an evolution of DCMD.
DCMD is an autosomal dominantly
inherited condition. However, thus far, the related genes and mutations have
not been identified. Previous linkage analysis showed that DCMD is associated
with the interval D7S493 to D7S526 at 7p15-p21[10-11]. We analyzed the haplotypes at the DCMD locus, and a
novel heterozygous RP
The human RP
Our study has a number of
limitations. Our findings revealed a case of a patient with DCMD carrying a
novel heterozygous RP
ACKNOWLEDGEMENTS
The authors are grateful to all
members in the family for their participation in the study.
Conflicts of Interest: Fu Y, None; Xie TH, None; Zhang
YL, None; Yang N, None; Shi XN, None; Gu ZH, None.
REFERENCES